Article on Genetic Clues in Parkinsonism: When Should You Suspect an Atypical Hereditary Syndrome?
M3 India Newsdesk May 19, 2025
This article outlines the genetic basis of Parkinson’s Disease, covering monogenic forms and related syndromes, and highlights the role of genetic testing in atypical or early-onset cases.

Parkinson’s disease is a neurodegenerative disorder of the central nervous system. Its cardinal features include a defined set of motor symptoms (bradykinesia, rigidity, tremor) as well as non-motor features like dementia, constipation, autonomic dysfunction, and psychiatric symptoms.
The pathological characteristic of PD is Lewy Bodies (formed by the aggregation of misfolded α-synuclein protein), which leads to the loss of dopaminergic neurons in the midbrain.
Besides age and environmental factors, genetic variation is estimated to contribute approximately 25% as a risk factor for PD. There are variants in single genes which are pathogenic, i.e. sufficient to cause disease. On the other hand, there are many common genetic variants which individually contribute to a small extent in the development of PD.
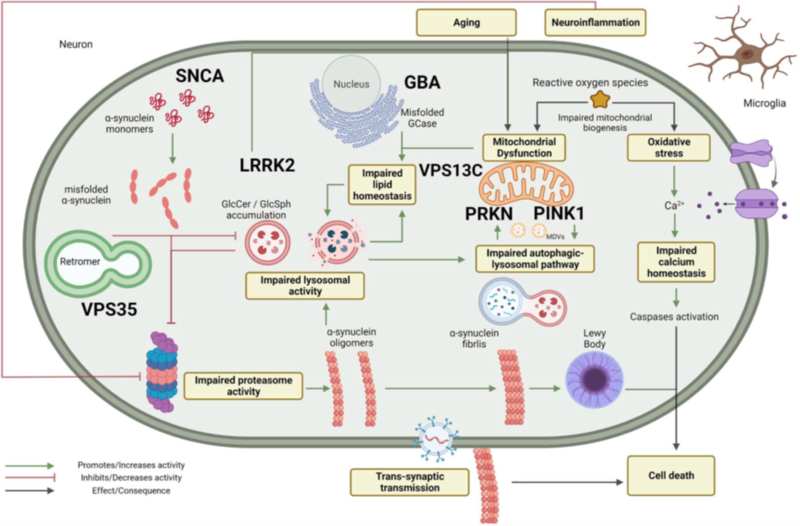
Figure 1: Schematic representation of the pathways involved in the pathogenesis of Parkinson’s Disease and the related genes.
This article will discuss in briefly discuss the common monogenic forms of PD as well as some genetic syndromes with overlapping PD features.
Case 1
- A 25-year-old male presented with progressive gait difficulty and bilateral hand tremors, evident since the age of 12 years.
- There were symmetric resting tremors in both hands, rigidity with lower limb dystonia and postural instability.
- Along with the above symptoms, the patient had also been experiencing hallucinations for the last 5 years.
- He had ideas of persecutory delusions, paranoid ideas, and auditory hallucinations, which did not respond well to psychotherapy. There was no family history of Parkinson’s Disease.
The MRI brain was normal, and so were the tests for Wilson’s Disease. Genetic analysis revealed the presence of a compound heterozygous mutation in the PARK2 gene. The patient was treated with pramipexole, amantadine & selegiline. However, his psychiatric symptoms reappeared after remaining stable for six months; hence, the dose of pramipexole was reduced, and clozapine was added to manage the psychiatric symptoms.
Autosomal Dominant Forms of Parkinson's Disease
- SNCA / α-Synuclein mutation (PARK1/4)
This was the first gene found to be associated with familial PD. Although the clinical phenotype is like that of sporadic PD, the affected patients have an earlier onset of symptoms (forties) and have more prominent autonomic dysfunction, hallucinations, psychiatric features, and cognitive impairment.
Certain atypical features like pyramidal signs, central hypoventilation and myoclonus can be seen. Autopsy reveals extensive Lewy Body deposition in the substantia nigra, limbic system, brainstem nuclei and the neocortex. PARK 1 is caused by point mutations in SNCA, whereas PARK 4 is due to triplication of the same gene, leading to an earlier onset and faster progression of symptoms.
- LRRK-2 / Leucine Rich Repeat Kinase -2 (PARK 8)
Mutations of the LRRK-2 gene are the most common genetic cause associated with autosomal dominant PD. It has a high prevalence in sporadic cases. The phenotype is like late-onset PD with a good response to Levodopa therapy. A “tremor dominant” phenotype is commonly observed, with later occurrence of falls and drug-induced dyskinesias. Non-motor features are less common and less severe than idiopathic PD. The clinical course is more benign compared to idiopathic PD. Pathologic variation is marked with “tau” aggregation, also noted in some cases.
- VPS35/ Vacuolar Protein Sorting 35 (PARK 17)
It is a rare disease-causing variant of PD seen in the Japanese & Caucasian population. The phenotype resembles typical late-onset PD.
Autosomal Recessive Forms of Parkinson's Disease
1. PRKN / Parkin (PARK 2)
It was the first gene linked to an autosomal recessive form of PD. It presents with an early-onset PD (age of onset < 40 years). Dystonia (commonly exercise-induced) is common in the initial stages; rest tremor is also common. There occurs unusual sensitivity to levodopa: dyskinesias and motor fluctuations are seen.
Psychiatric features (depression, suicidal tendencies, psychosis) precede the onset of PD features. There is symmetric motor impairment, which corresponds to symmetrically reduced striatal tracer uptake in both hemispheres. Autonomic affection is present, whereas cognition is usually intact. The clinical course is usually benign.
2. PINK1 Mutation (PARK 6)
It is the second most common cause of early-onset PD. Clinical features resemble those of typical PD with unilateral onset tremor, bradykinesia, and rigidity with a good response to levodopa. In some instances, psychosis, behavioural disturbances, and a frontal-type dementia phenotype have been observed.
3. DJ-1 Mutation (PARK 7)
It is a rare cause of autosomal recessive early-onset PD. Clinical features are like Parkin and Pink1 mutations.
4. ATP13A2 Mutation (PARK 9)
Also known as Kufor-Rakeb Syndrome. It presents with a combination of extrapyramidal and pyramidal features with incomplete supranuclear vertical gaze palsy. Other features include presence of oculogyric dystonic spasms, autonomic dysfunction and facial–faucial–finger mini-myoclonus. Visual hallucinations and dementia can also be present. In some cases, there may be some clinical overlap with neuronal ceroid lipofuscinosis (NCL).
5. PLA2G6 Mutation (PARK 14)
- It is also classified as NBIA (PLAN). The salient clinical features of early onset include motor and mental retardation, prominent early onset cerebellar ataxia, truncal hypotonia, pyramidal signs and optic atrophy.
- The dystonia-parkinsonism phenotype is a relatively late-onset form of this mutation; these patients are very sensitive to levodopa with early-onset dyskinesias.
- There can be associated oculomotor abnormalities, cognitive impairment, and psychiatric features. Brain MRI reveals cerebellar atrophy; iron deposition is often absent in the dystonia-parkinsonism phenotype.
Other Genetic Syndromes Overlapping with Parkinson's Disease
-
GBA1 gene
The GBA1 gene encodes for β-Glucosylceramidase; biallelic mutations in the GBA1 gene result in Gaucher’s Disease, a lysosomal storage disorder. More recently, this gene has been linked to the development of PD. Although the clinical features are like idiopathic PD, GBA1-PD is characterised by earlier onset of symptoms, rapid progression with worse motor impairment and higher chances of autonomic and cognitive dysfunction. Psychiatric features like impulsive-compulsive behaviour, hallucinations and anxiety are also seen.
- DCTN1 Mutation (Perry Syndrome)
The DCTN1 gene encodes for Dynactin, a part of a motor complex associated with axonal transport. Four important clinical features of this syndrome include Parkinsonism, depression/apathy, central hypoventilation, and weight loss. The PD features include a young onset, rapidly progressive disease, with more symmetry of clinical features compared to sporadic PD.
Other features of this syndrome include oculomotor signs, frontal affection (hyperorality, disinhibition & dysexecutive syndrome), cognitive impairment and sleep disorders. Brain pathology includes TDP-43 inclusions in the basal ganglia and brainstem with degeneration in the substantia nigra.
Case 2
- An 18-year-old male presented with unilateral right-hand tremors for the last 10 months associated with gait difficulties.
- The tremors were present only at rest; no aggravating or relieving factors were observed.
- There was no significant family history; he did not adequately respond to Propranolol, which was prescribed by the primary care physician.
Examination
The patient had an expressionless face with reduced eye blinking. His vertical saccadic eye movements were slow. Rigidity and bradykinesia were present in the limbs along with rest tremors in the right hand. Gait imbalance was present with swaying to the right side.
Multiple investigations, including routine blood profile, Wilson’s, metabolic profile, and MRI of the brain, were unrewarding. Considering a profile of young-onset Parkinsonism, he was subjected to genetic testing, which revealed the presence of a homozygous deletion in the ATP13A2 gene; hence, the diagnosis of Kufor-Rakeb Syndrome (PARK-9) was confirmed. He was started on a low dose of Levodopa/Carbidopa with mild improvement in tremors and walking.
Conclusion
With improving understanding of the pathogenesis of Parkinson’s Disease, a greater number of genes are being discovered that are being directly or indirectly implicated in the same. In the future, gene-directed therapy is likely to emerge, which will supplement the already available symptomatic therapies for Parkinson’s disease. Hence, the clinician should be aware of the atypical clinical features which might point towards a genetic cause of PD and offer genetic testing when indicated.
Disclaimer- The views and opinions expressed in this article are those of the author and do not necessarily reflect the official policy or position of M3 India.
About the author of this article: Dr. Annesh Bhattacharjee is a Consultant Neurologist at Cosmo Medical, Guwahati.
-
Exclusive Write-ups & Webinars by KOLs
-
 Daily Quiz by specialty
Daily Quiz by specialty -
 Paid Market Research Surveys
Paid Market Research Surveys -
Case discussions, News & Journals' summaries
